
Presentado por:
Dra. Mirtha Briñez Juarez
Servicio de Hematología
Dr. Rolando Hernández Pérez
Servicio de Dermatología
Hospital General "Dr. Luis Razetti"
Barinas/Venezuela
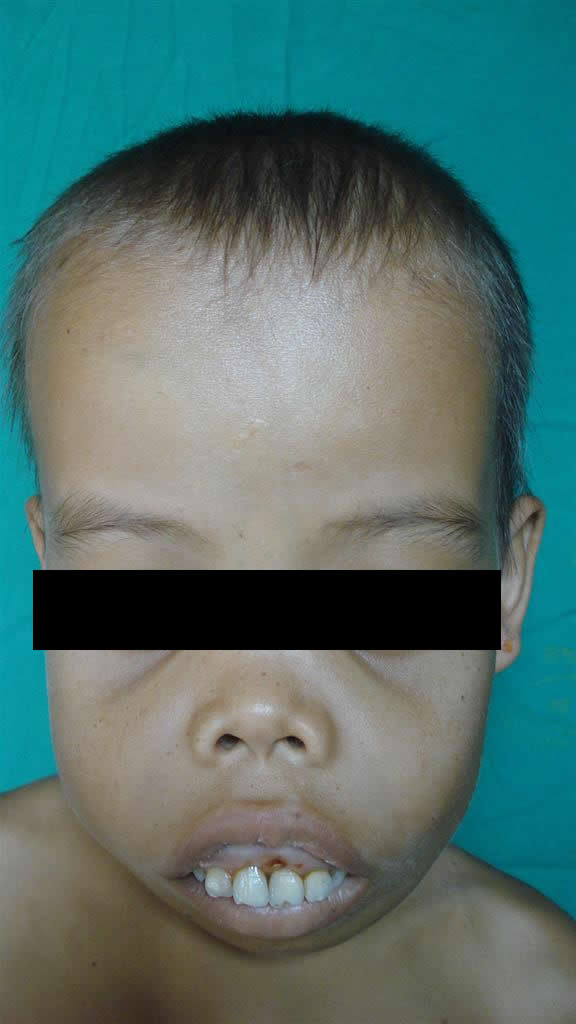
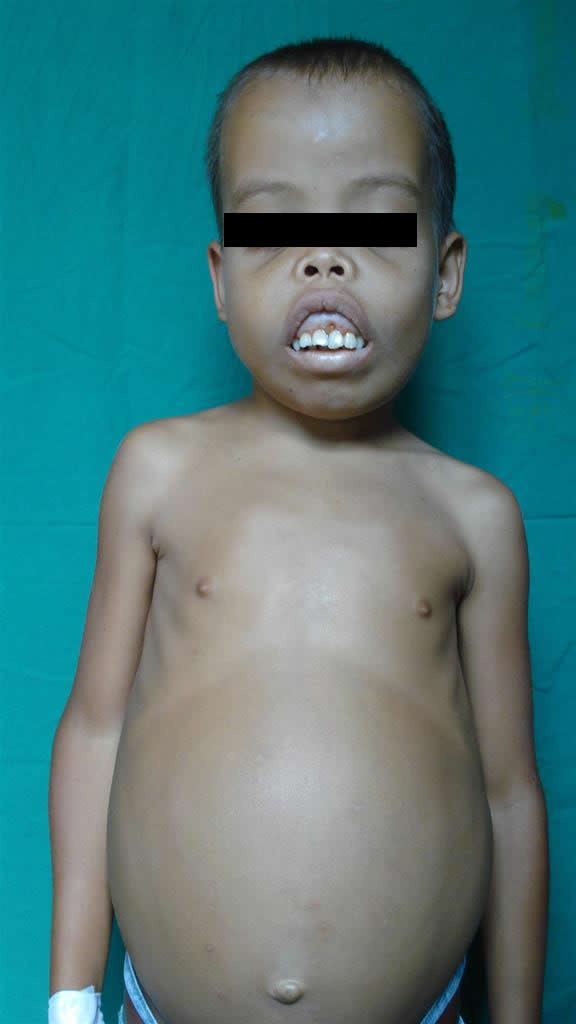
Paciente masculino de 13 años de edad, en control irregular por el servicio de hematología desde el año 2005. Al ingreso color cetrino (amarillo-verdoso) en piel, mucosas pálidas, petequias en mucosa gingival; deformidad de huesos de cara y cráneo: aplastamiento de tabique nasal, protrusión de frontales, malares y maxilar superior, con prominencia de las regiones acrales de los mismos, hepato-esplenomegalia importante, déficit pondo- estatural y desnutrición moderada.
Laboratorio
Electoforesis de Hb: A2 F
Fetal: 62% – A2: 38% – A:0 %
En controles maneja cifras de Hb: 6.5Gr%. Compensado hemodinamicamente pese a cardiomegalia. No se somete a régimen de transfusión por ser portador de Ac. Irregular (anticuerpos irregulares no detectados aún) y no disponer de tratamiento para quelación de hierro.
Se solicitó interconsulta con Dermatología: se observa una piel seca, tensa, verde amarillenta, discretas máculas hipercrómicas puntiforme; boca permanentemente abierta, xerostomía y púrpuras en mucosa gingival.(Favor ver foto clínica N° DSCO1844.JPG-DSCO1845.jPG-DSCO1843.JPG)
 |
 |
 |
| DSCO1844.JPG | DSCO1845.jPG | DSCO1843.JPG |
Frotis: anisocitosis, anisocrómia, poiquilocitosis y policromasia.(Favor foto de frótis periférico: HLR-BNS143.jpg -HLR-BNS142.jpg)
 |
 |
| HLR-BNS143.jpg | HLR-BNS142.jpg |
Se concluye en una TALASEMIA MAYOR MAL CONTROLADA
Comentarios:
Las TALASEMIAS son anemias hereditarias que ocurren por mutaciones que afectan la síntesis de la hemoglobina.
Clasificación
|
1)TALASEMIA β grave Anemia de Cooley) Talasemia mayor Talasemia intermedia |
Anemia grave, retraso del crecimiento, hepatoesplenomegalia, expansión de la médula ósea y deformación ósea. Dependiente de transfusiones. No requiere transfusiones regulares. |
|
2) Carácter de Talasemia (α o β |
Anemia leve con micrositosis e hipocromía. |
|
3) Enfermedad Hb H (tal α) |
Anemia hemolítica moderadamente grave, ictericia y esplenomegalia. |
|
4) Hidropesía fetal (tal α) |
Muerte in utero por anemia grave. |
|
5) Portador silencioso (α o β) |
Hematológicamente normal |
α: alfa
β: beta
En la TALASEMIA β hay deficiencia en la síntesis de la globina β, en tanto que en la TALASEMIA α la síntesis de la globina α es deficiente. La disminución de la síntesis de uno o dos polipétidos globina origina un deficiencia de la acumulación de hemoglobina , que origina eritrocitos hipocrómicos y microcíticos. Estas anormalidades de los eritrocitos son las características más constantes y distintivas de este grupo de trastornos. La incidencia y prevalencia de estos trastornos son muy variable. El más común es el Carácter de Talasemia una anemia insignificante en la clínica, leve; es muy común en algunas partes del mundo. El Carácter de la Talasemia suele representar la forma heterocigota de TALASEMIA α y β . En consecuencia cuando en Caracte de de Talasemia es común, homocigoto, se encontrarán con mayor frecuencia pacientes con afección más grave. En los EEUU la frecuencia de Talasemia β es más alta en grupos étnicos de origen mediterráneos y de algunas partes de África y de Asia, en tanto que la Talasemia α es más común en personas del Asia. En los EEUU se conocen alrededor de 5000 personas con la forma grave de Talasemia. En nuestro país no hay registro serio de Talasemia.
TALASEMIA β GRAVE (Anemia de Cooley) (aparentemente nuestro caso)
La Talasemia β grave ocurre en pacientes homocigotos de mutaciones que conducen a una disminución de la síntesis de globina β. Como se afectan ambos genes de globina β, hay una deficiencia notable de su síntesis. Pero la producción de globina α continúa en un índice casi normal.
Clínicamente al nacer estos pacientes con TALASEMIA MAYOR son casi normales hematológicamente, ya que la síntesis de globina γ es normal y la producción de HB F (hemoglobina fetal) es en consecuencia adecuada. Sin embargo, a medida que se termina el cambio de Hb F a Hb A durante el primer año de vida, la defieciencia de producción de globina β se torma obvia. Hacia los 6 a nueve meses de edad los padres llevan al lactante al médico por anemia grave, que se manifiesta por palidez, deficiencia en el crecimiento e ingestión inadecuada de alimentos y en ese momento el examen revela la presencia de hepato-esplenomegalia notable. La Hb puede ser de 3-6 g/100 ml.y los eritrocitos muestran una microcitosis , hipocromía, y fragmentación grave. El estudio de la sangre del lactante demustra una ausencia de Hb A o valores bajos.
Antes del uso regular de las transfusiones sanguíneas, estos niños se deformaban notablemente por la expansión de los espacios medulares del cráneo (espacios de punta de pelo). La osteoporosis grave causa fracturas patológicas y la anemia debilidad e inanición, la muerte era común hacia los dos o tres años de edad. Si los pacientes son tratados regularmente y precozmente con transfusiones sanguíneas prácticamente el niño no manifiesta lesiones clínicas óseas o estigmas. Como el hombre tiene una capacidad muy limitada para eliminar el hierro, las transfusiones sanguíneas regulares provocan de manera inevitable una acumulación importante de hierro. Por eso es importante la utilización de Quelantes del hierro en estos pacientes tempranamente.
Lamentablemente nuestro paciente acude a la consulta tardíamente, y no tiene posibilidades económicas, operativas, ni culturales para cumplir con la conducta en estos casos. Además hay una situación social y psicológica familiar en desventaja para este pequeño.