
 Estimado Félix:
Estimado Félix:
Te envío unos comentarios recientemente publicados en el NEJM, (2007), Nature Reviews Cancer (2006), Nature (2007), Nature (2007), por un grupo de investigadores del Programa de Oncología Molecular del Centro de Investigación Nacional del Cancer, en Madrid (CNIO), España, así como por otros autores que trabajan en la misma línea de investigación, sobre el papel del envejecimiento celular, del p53 y de la regresión tumoral en la carcinogénesis.
Me parecieron de gran importancia y actualidad si se toma en cuenta como la presencia, disminución o ausencia del p53 juega un importante papel en el desarrollo de lesiones tumorales pre-carcinomatosas y carcinomatosas cutáneas y de otros órganos y sistemas y como la presencia de la inmunidad innata o natural, tan bien descrita en tu reciente comentario en Inmunodermatología del 18 de Mayo de 07, e impulsada por Charles Janeway Jr, juega importante papel en los fenómenos de regresión tumoral.
Si lo crees conveniente, puedes incluirlo en tu columna de Inmunodermatología. Como siempre, considérate en plena libertad y confianza para de mutuo acuerdo, hacer las modificaciones que creas conveniente. Sabes que soy muy receptivo a cualquiera sugerencia de tu parte.
Espero por tu respuesta.
Un abrazo, Guillermo Planas G.
“PAPEL DEL “ENVEJECIMIENTO CELULAR”, DEL P53, Y DE LA REGRESION TUMORAL EN LA CARCINOGENESIS”
Recientemente, Serrano y Collado, investigadores del Programa de Oncología Molecular del Centro de Investigación Nacional del Cáncer, en España (CNIO), Madrid, publicaron un artículo de máximo interés en Nature Reviews/Cancer (1), sobre el “envejecimiento celular”, donde explican éste interesante fenómeno, inducido por oncógenos (OIS), el cual ocurre durante los estadios tempranos de la tumorogénesis
Refieren los autores que una de las vías de mayor relevancia en cuanto a “stress” celular es derivada de los signos proliferativos aberrantes de oncógenos, los cuales pueden eventualmente disparar el envejecimiento a través de un proceso conocido como “oncogene-inducid-senescence” (OIS).
El envejecimiento es una respuesta celular ante el “stress”. El concepto es aplicado en general a la interrupción de proliferación irreversible de células causadas por varios “stress” (daños oxidativos incluyendo disfunción del telómero, daño del DNA, varias drogas quimioterápicas: dexosirubicina, cisplantin, vincristina, etc y las OIS. (Ver Fig. 1, que se explica por si misma).
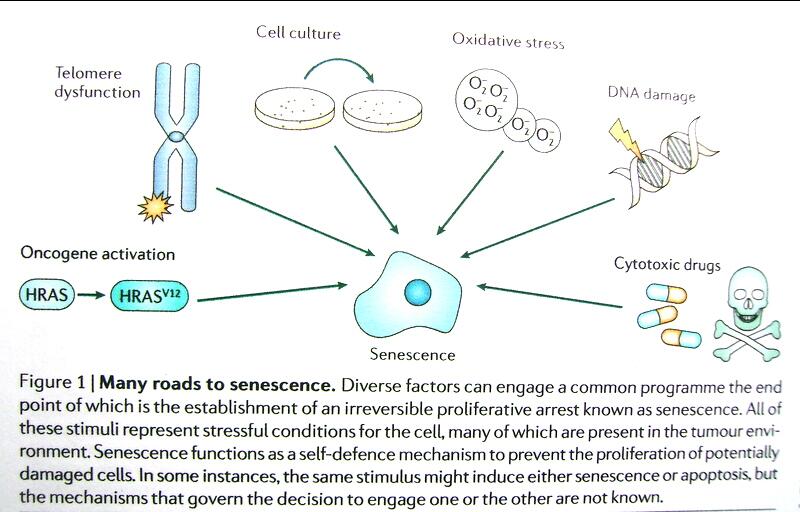 Figura Nº. 1:Muchas vías hacia el envejecimiento. Tomado del artículo de Collado M & Serrano M. The power and promise of oncogene induced- senescence markers. Nature Reviews/ Cancer (1)
Figura Nº. 1:Muchas vías hacia el envejecimiento. Tomado del artículo de Collado M & Serrano M. The power and promise of oncogene induced- senescence markers. Nature Reviews/ Cancer (1)
Estos investigadores demostraron que las células tumorales “envejecidas” son abundantes dentro de lesiones neoplásicas premalignas y escasas en tumores malignos (Figura 2) y que éstos eventos abren las puertas para la utilización de “marcadores de envejecimiento” con fines diagnósticos y pronósticos, así como una vía para determinar, mediante dichos marcadores, los efectos de la quimioterapia sobre células envejecidas, cuando se realizan protocolos para tal fin.
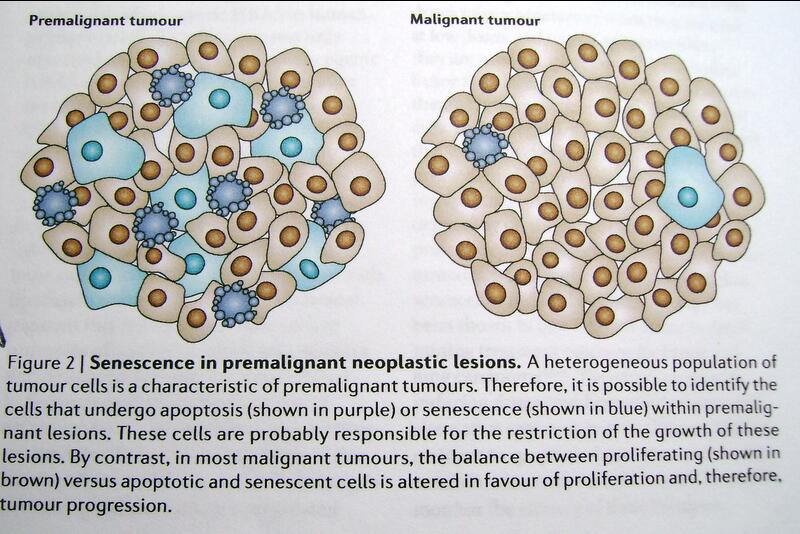 Fig. Nº. 2: Envejecimiento en lesiones neoplásicas premalignas y malignas tomado del artículo de Collado M & Serrano M. The power and promise of oncogene-induced senesence markers . Nature Reviews/ Cancer (1)
Fig. Nº. 2: Envejecimiento en lesiones neoplásicas premalignas y malignas tomado del artículo de Collado M & Serrano M. The power and promise of oncogene-induced senesence markers . Nature Reviews/ Cancer (1)
En la misma línea de investigación, Serrano M, publica en Mayo de 2007 en el NEJM, en la sección “Clinical Implications of Basic Research”, bajo el título de “Cancer Regressión by Senescence” (2) interesantes conclusiones basadas en investigaciones propias y de otros autores que trabajan sobre la misma materia-Xue W, et al. (3); Ventura A et al. (4); Martins CP et al. (5); Collado M & Serrano M. (1)- sobre el papel que juega la proteina p53 en éstas alteraciones que sufre la célula cancerígena en el proceso de tumorogénesis y los efectos del envejecimiento sobre la regresión tumoral.
Pero antes de incursionar sucíntamente en éstos interesantes trabajos, es importante definir brevemente lo que se conoce por envejecimiento celular, su relación con la apoptosis y el papel que juega el p53 en los eventos de carcinogénesis, especialmente en tumores cutáneos frecuentes.
La “senescence” o senectud o envejecimiento celular es un proceso mediante el cual la célula responde ante un “stress”, que supone un freno en la proliferación celular, pero no necesariamente induce a la célula a la muerte celular programada (Apoptosis), más aún, éstas células pueden permanecer metabólicamente activas.
En la década de los años 60, las investigaciones de Leonard Hayflick y Paul Moorhead marcaron pauta con el descubrimiento que las células humanas derivadas de tejido embriónico, se pueden dividir un número finito de veces en cultivo. De esos experimentos nació el fenómeno conocido como límite de Hayflick, o fase III del fenómeno de multiplicación de las células en cultivo, conocida también como “envejecimiento replicativo” (ER) o detención de la división celular a un nivel donde la célula puede o no morir.
Posteriormente en la década del 90, estos autores trabajaron con fibroblastos y otras células donde se ha comprobado el ER como queratinocitos, células endoteliales, linfocitos, células adrenocorticales, células del músculo liso vascular, condrocitos, etc. y en muchas especies de animales como ratones, pollos, galápagos, tortugas, demostrándose relación entre el número de células que se replican en cultivo y la longevidad de la especie. P.ej. las células de las tortugas pueden vivir cerca de cien años, dividiéndose alrededor de 110 veces, en tanto que las del ratón se dividen 15 veces.
El p53 es un gen localizado en el brazo corto del cromosoma 17 entre las bandas 17p12 y la 17p13.8. La mutación en el gen supresor, denominado por algunos autores “el guardián del genoma” por su desempeño fundamental como supresor tumoral, constituye la alteración génica más frecuente en la mayoría de neoplasias humanas con deleción en p53, siendo marcadores en procesos tardíos de carcinogénesis (6). Por ello es que la función de p53 inactivada en ciertas neoplasias, puede estar implicada, como señalamos antes, en la progresión tumoral, más que en la transformación maligna. Se ha relacionado a las metástasis con la supresión completa de p53.
Son conocidos los trabajos que destacan el papel del gen p53 en el desarrollo de tumores frecuentes de la piel como el Carcinoma Basocelular (CBC) y el Carcinoma Espinocelular (CEC). (7)
Las mutaciones del p53 se han demostrado en cerca de la mitad de los CBC -Bolhakov S et al. (8); Rady P et al. (9); Ziegler A et al. (10)- resaltando el papel de la radiación solar, como mutágeno, no obstante, otro autores como Gailani MR et al. (11) han encontrado que es muy infrecuente que en el CBC se produzca deleción del p53. Además éste último autor y sus col. analizaron 18 tumores, de los cuales en 11 CBC (61%) se observó pérdida de los marcadores del ADN 9q y en 11 (61%), descubrieron mutaciones del p53 y en 7 (39%), se observaron alteraciones de ambos genes.
En síntesis en CBC están involucrados dos genes supresores: PTCH, el cual es el homólogo humano del gen patched de la Drosofilia (PTC en ratones, gen que identificaron en el Síndrome de Gorlin: Síndrome del Nevus Basocelular, como lo describieron los investigadores Han H et al. (12) y Johnson RL, et al (13).
Al igual que en el CBC, en el CEC, sucede un mecanismo similar, inducido por la LUV, que daña el ADN celular. La inducción de la expresión del gen p53, se produce como consecuencia de las rupturas de las cadenas de ADN. La P53 estimula la expresión de p21 que bloquea la progresión del ciclo celular en G2 al unirse e inhibir las cinasas dependientes de la ciclina (CDK) 2 y 4. La inhibición de la progresión del ciclo celular, permite la reparación de ADN, antes que se replique en la fase S, para evitar que se mantengan las mutaciones que se han producido. Si el daño es severo, se produce Apoptosis.que está mediada por inducción de BAX por el p53. Si el gen p53 se inactiva a causa de la mutación o deleción, no se producirá el bloqueo del ciclo celular, ni apoptosis, como respuesta al daño de ADN. genómico, las células lesionadas persistirán y podrán sufrir expansión clonal y por tanto formación de tumores.
Serrano sostiene en su reciente artículo (2), que los cánceres en ratones pueden ser eliminados a través de la activación de un gen singular, TP53, el cual contiene la proteina p53. Se piensa que la p53 es razonablemente el más importante sensor de “Stress” que poseen los mamíferos. Bajo circunstancias normales, ésta proteina es de poca importancia por la rapidez en su degradación, pero casi cualquier tipo de “stress”, incluyendo daño en el ADN y señalamiento oncogénico, detiene la degradación de p53 y afecta su activación.
Señala el autor que los tumores están en un ambiente altamente estresante que impone fuerte presión para eliminar el sistema protectivo mediado por p53. Casi todos los cánceres humanos, tienen algún tipo de daño en la vía de p53 (50% tienen mutaciones inactivantes en TP53 o se asocian a algún tipo de deleción). Tal vez la mayor sorpresa obtenida en estos tres estudios (3, 4, 5), fue el descubrimiento de envejecimiento como mecanismo de regresión tumoral. En los casos de linfomas inducidos por radiación, la regresión sucedía rápidamente, asociada a abundante apoptosis. En contraste, los hepatocarcinomas y sarcomas, regresaban más lentamente, sin signos de apoptosis.
Estos hallazgos condujeron a los investigadores a determinar si el p53 conducía a envejecimiento tumoral. Como se mencionó anteriormente, el envejecimiento, así como la apoptosis, son respuestas al “stress”, pero incapaces a inducir a la muerte celular, pero si provocan un freno permanente en la proliferación celular. El factor de que las células envejecidas se encuentren en alta proporción en lesiones pre-malignas (1) (Fig. 2) es consistente con el concepto de que los tumores se encuentran en un ambiente altamente estresante y que el envejecimiento puede constituir un eficiente freno a la progresión tumoral.
Xue y col (3), en 2007, investigaron las bases histopatológicas de la “regresión tumoral”.Los autores, propusieron que la activación del p53 induce envejecimiento e involución tumoral y que resultaba sorprendente, ya que se concebía al envejecimiento como un programa citostático. Realmente las células transformadas acumularon actividad SA-b galactidasa, marcador del envejecimiento in vitro e in vivo (1), pero subsecuentemente permanecían detenidas, siguiendo a la reactivación del p53 “in vitro”, sugiriendo que la regresión tumoral, conforma un proceso autónomo no celular.
Los autores, describieron microscópicamente (3), tumores producidos en diferentes etapas, seguidos de la reactivación de p53, en los cuales demostraron una reacción inflamatoria progresiva que comprometía a los leucocitos polimorfonucleares, inicialmente en las regiones peri-tumorales y posteriormente a través del tumor. Se observó también un intenso infiltrado peri-vascular en tumores en regresión, que conducían a una eventual pan-vasculitis, caracterizada por vasos esclerosados, hemorragia y eritematofagocitosis. Los análisis morfológicos, con IF y citometría de flujo, identificaron los leucocitos infiltrantes como neutrófilos, macrófagos y células “natural killer”. Estos hallazgos histopatológicos, soportan un modelo de eventos secuenciales, iniciados por la reactivación de p53 en los tumores, activación de una respuesta inflamatoria dramática, seguida por destrucción de las células tumorales y neovascularización.
Las células envejecidas adquieren un señalamiento de expresión genética que incluye sobreregulación de citocinas inflamatorias y otros moduladores inmunes.
En el estudio de Yue y col, se identificó un nuevo mecanismo de supresión tumoral que compromete interacciones cooperativas entre un programa de células tumorales envejecidas y el sistema inmune innato, demostrando además la capacidad que tienen las células envejecidas de “regresar” in vivo a pesar de la naturaleza citostática del programa de envejecimiento. Si tal “regreso” es un rasgo general del envejecimiento in vivo no está claro, pero cuando está presente, puede reforzar la acción de supresión tumoral del envejecimiento en casos premalignos o aquellos casos siguiendo tratamiento de tumores con terapias que promuevan el envejecimiento o diferenciación.
Los resultados de Xue y col. identificaron un escenario en el cual el sistema inmune innato es inducido a atacar coordinadamente a las células tumorales presumiblemente a través de la fagocitosis y muerte citotóxica directa, facilitando su eliminación.
Aunque se ha establecido que la inflamación crónica inducida por células envejecidas del estroma y otros factores, promueven la tumorogénesis, las investigaciones de estos autores, ilustran como las células inmunes innatas, cuando son dirigidas contra las células envejecidas, pueden tener también efectos antitumorales.
Los tres estudios, en los que se ha hecho énfasis (3, 4, 5), soportan las investigaciones hechas con drogas dirigidas a activar el p53 e indican que el envejecimiento en adición a la muerte celular, constituyen hechos relevantes en terapias anticáncerígenas.
Referencias:
1) Collado M & Serrano M. The power and the promise of oncogene-inducid senescence markers. Nature Reviews Cancer. 2006; 6:472-476
2) Serrano M. Cancer Regression by Senescence. Clinical Implications of Basic Research. NEJM, 2007 Number 19; 356:1996-1997
3) Xue W, Zender L, Miething C, et al: Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature 2007;445:656-660
4) Ventura A, Kirsch DG, McLaughlin ME, et al. Restoration of p53 function leads to tumour regression in vivo. Nature 2007;455:661-665
5) Martins CP, Brown-Swigart L, Evan GI. Modeling the therapeutic efficacy of p53 restoration in tumors. Cell 2006; 127:1323-1334
6) Baker. Supression of human colorectal carcinoma cell growth by wildtype p53. Science 1991 b;249:912-15
7) Dans M & Fakharzadeh SS. Bases Genéticas del Cáncer de la Piel (en) Cáncer de la Piel ( Eds) Rigel DS Friedman RJ, Dzubow LM, Reintgen DS, Bystryn JC, Marks R. Elsevier España. SA, Génova, 17.3º, 28004 Madrid. España, 2006: 15-27
8) Bolshakov S, Walter CM, Strom SS, et al. p53 mutations in human aggressive and nonaggresive basal and squamous cell carcinoma. Clin Cancer Res 2003; 9:228-234
9) Rady P, Scinicariello F, Wagner RF Jr, et al. p53 mutations in basal cell carcinomas. Cancer Res 1992 ; 52 :3804-3806
10) Ziegler A, Leffel DJ, Kunala S, et al. Mutation hotspots due sunlight in the p53 gene of nonmelanoma skin cancers. Proc Natl Acad Sci 1993; 90:4216-4220
11) Gailani MR, Leffell DJ, Ziegler A, et al. Relationship between sunlight exposure and a key genetic alteration in basal cell carcinoma. J Natl Cancer Inst 1996; 88:349-354
12) Hahn H, Wicking C, Zaphiropoulous PG, et al.Mutations of the human homolog of Drosophila patched in the nevoid basal cell carcinoma syndrome.Cell 1996; 85:841-851
13) Johnson RL, Rothman AL, Xie J, et al. Human holmolog of patched , a candidate gene for the basal cell nevus syndrome, Science 1996; 272:1668-1671