
Mujica Raiza (Residente de segundo año), Houda Drikha (Dermopatologa, jefe del servicio), Estrada Pedro (Genetista),
Universidad Centroccidental “Lisandro Alvarado”, Postgrado en Dermatología,
Hospital Central “Dr. Antonio Maria Pineda”. Barquisimeto – Venezuela.
La eritroqueratodermia simétrica progresiva o Síndrome de Gottron, es una genodermatosis de prevalencia muy baja, genéticamente heterogénea, y la mayoría de los casos ocurren de forma aislada, su causa es una mutación en el gen de la loricrina ubicada en 1q36, la descripción de la enfermedad data de 1891 y la realizó Darier, mientras que Gottron acuño el termino en 1922, se trata de un trastorno de la queratinización con una proliferación epidérmica aumentada, las manifestaciones clínicas generalmente empiezan durante la infancia temprana con el desarrollo de placas hiperqueratósicas policíclicas sobre una base eritematosa, cubierta a su vez por descamación fina.
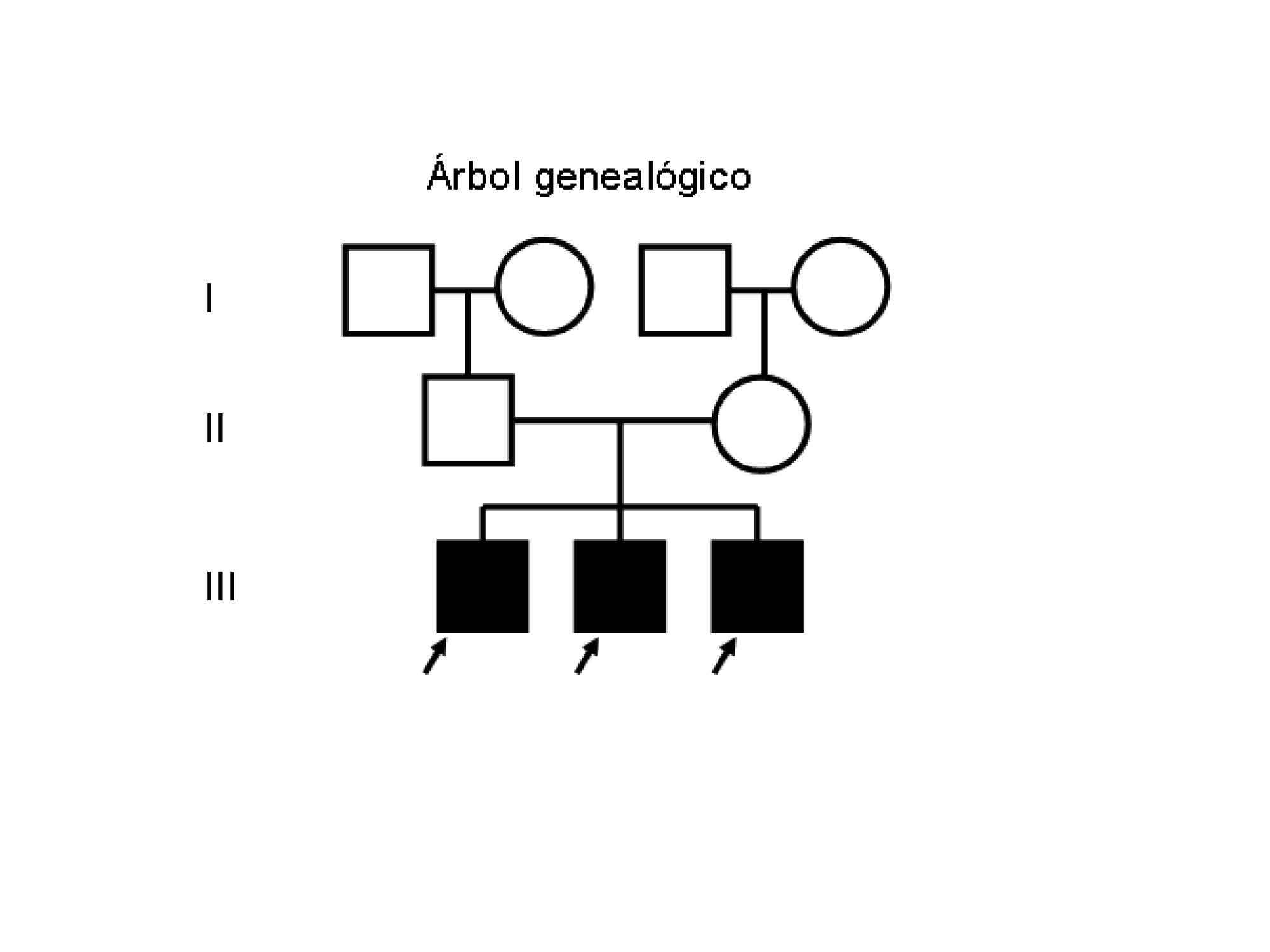
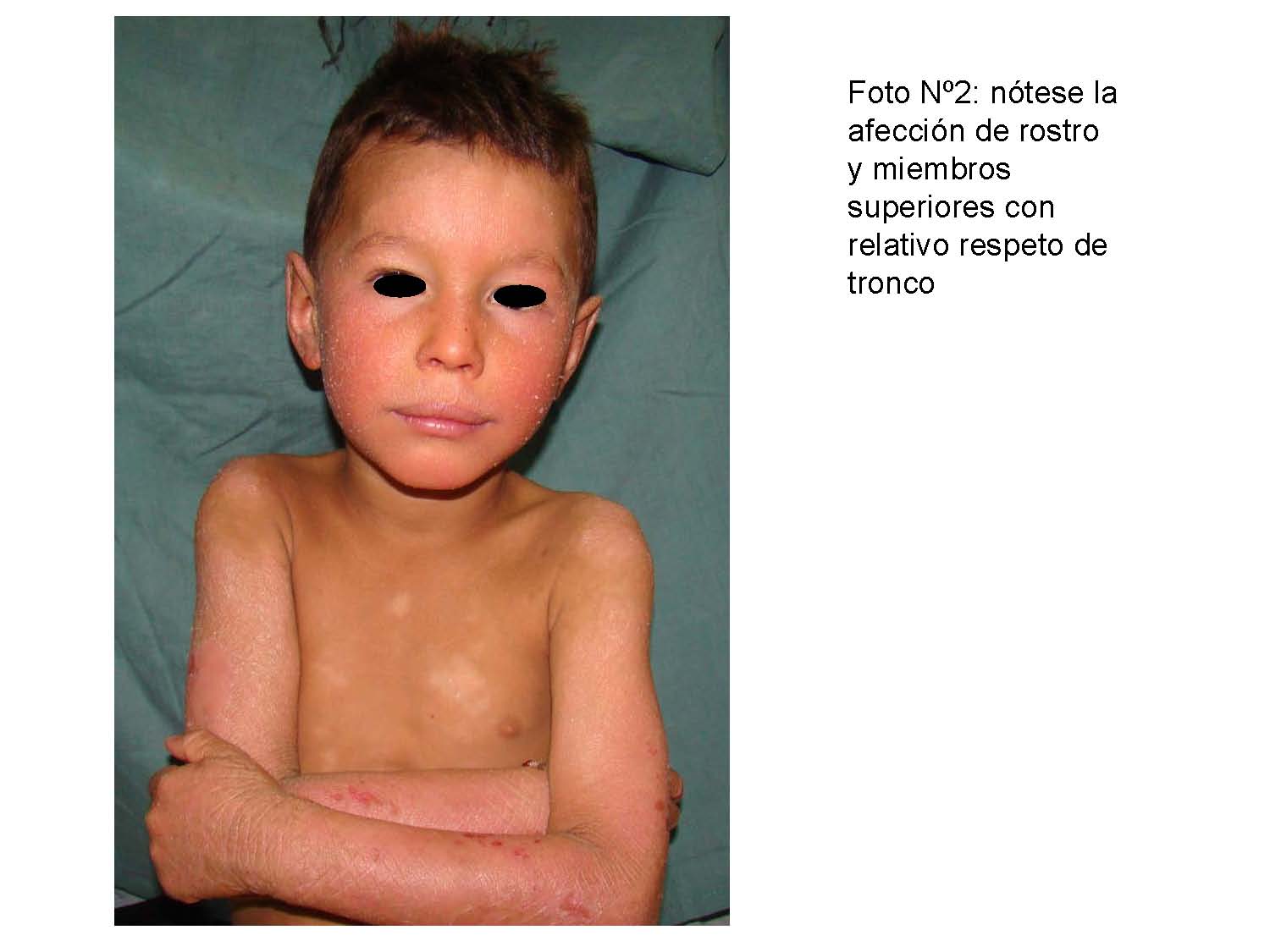
Al servicio de Dermatología del Hospital Universitario “Dr. Antonio María Pineda”, Barquisimeto acudieron tres hermanos, todos masculinos, de diez, ocho, y cuatro años respectivamente, naturales y procedentes del medio rural, con inicio de enfermedad actual a los tres meses, dos meses y al mes de vida respectivamente, dada por “escamas, resequedad” y dificultad para la plegabilidad de la piel, que aparece inicialmente en manos y pies de manera simétrica, posteriormente se diseminan a antebrazos, brazos y región proximal de tronco así como rostro y región glútea.
Antecedentes personales patológicos: no contributorios
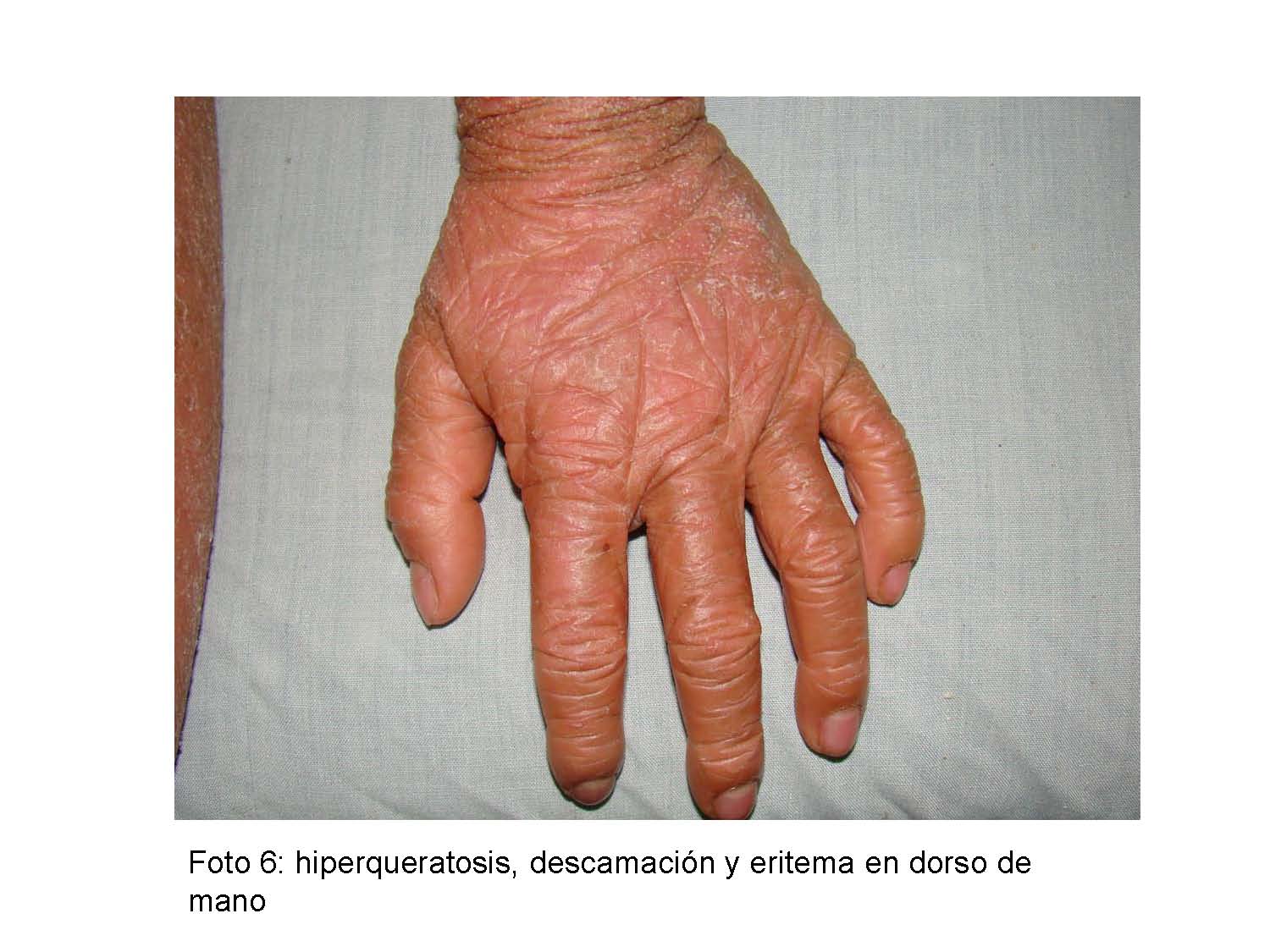
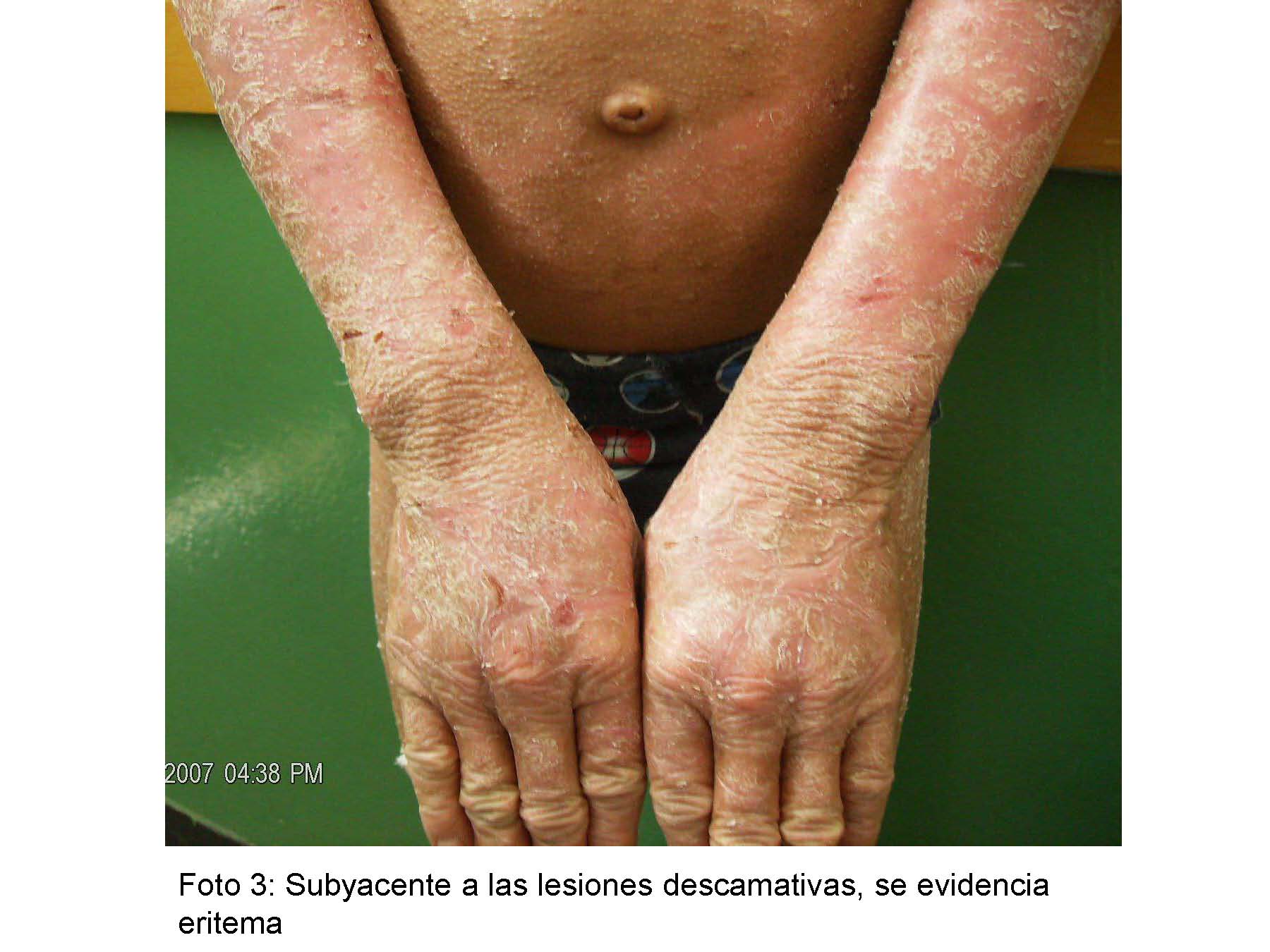
Al examen físico pacientes en buenas condiciones generales, activos, fototipo cutáneo II/VI, normotónicos, placas hiperqueratósicas, apergaminadas, de difícil desprendimiento con descamación furfurácea que asientan sobre una base eritematosa, alternando con sectores de liquenificación, ubicadas de manera simétrica en ambos miembros inferiores y superiores, región glútea y rostro, hiperqueratosis palmo plantar y uñas convexas, evaluación psicomotor normal
Paraclínicos
Hematología completa, urea, creatinina, TGO. TGP, Glicemia, amilasas séricas, uroanálisis, coproanálisis y ecosonograma abdominal, resultaron todos dentro de límites normales
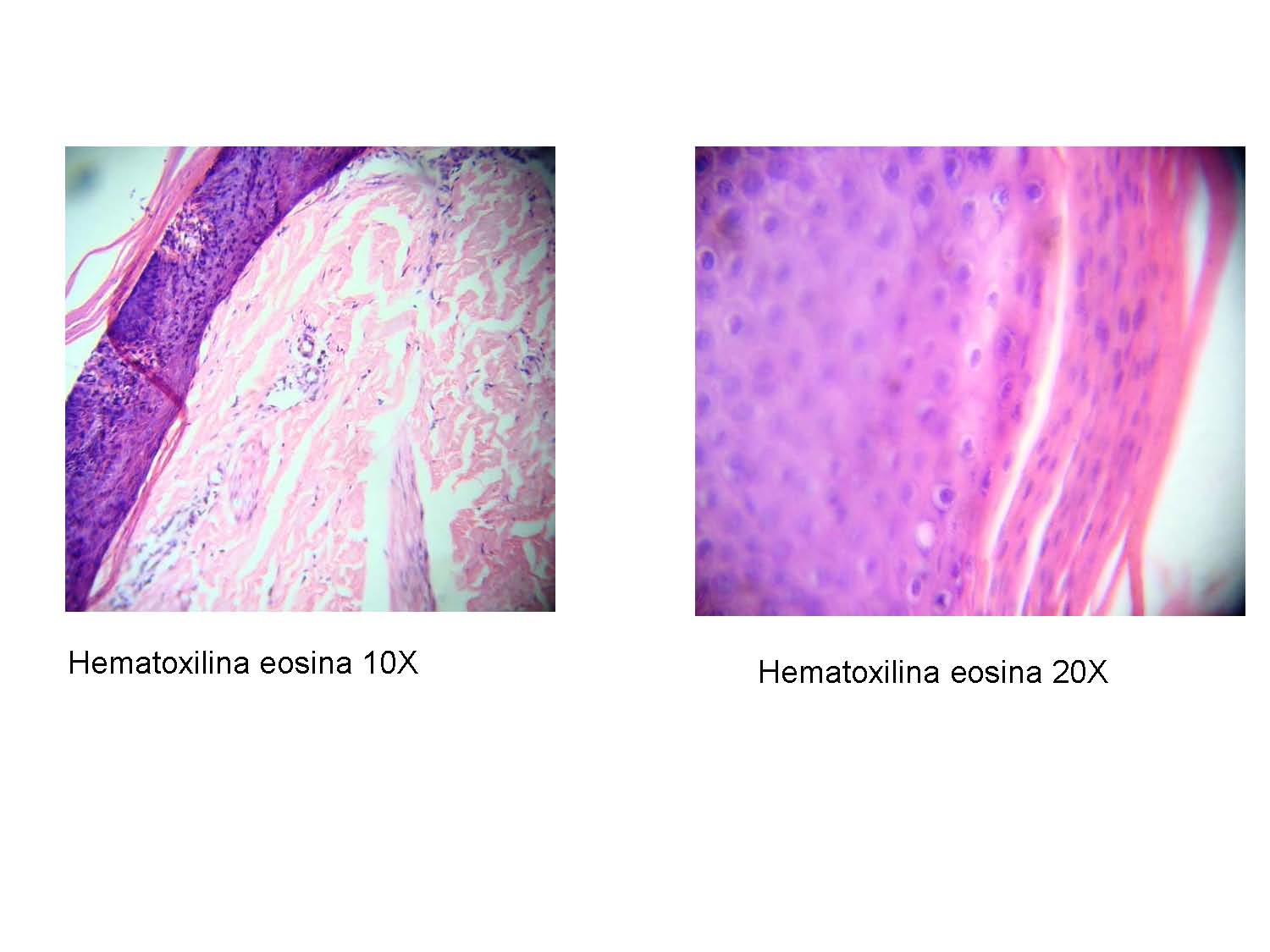
Estudio histológico reportó: epidermis con sectores de paraqueratosis los cuales subyacentemente presentan hipogranulois, disqueratois y espongosis, dermis de aspecto papilomatoso, con infiltrado linfocitario perivascular escasos neutrófilos
Desde el punto de vista clínico e histopatológico los tres casos fueron diagnósticados como eritroqueratodermia simétrica progresiva
Se solicito interconsulta con el servicio de genética quienes informaron que: el patrón de herencia en esta enfermedad es variable, son los primeros casos reportados en su árbol genealógico por lo que se trata de una neomutación y en los casos propósitos se plantean la posibilidad de que se trate de un patrón de herencia recesiva ligada al X o un mosaicismo germinal.


Actualmente reciben tratamiento con Fórmula enriquecida con aceite de Karité (10%), formulas tópicas compuestas por urea al 10%, glicerina, parafina liquida, glicina y tocoferol, protección solar con preparados que contienen Mexoryl®, logrando discreta mejoría del cuadro clínico.
Es importante reseñar que la eritroqueratodermia simétrica progresiva, es una genodermatosis de muy baja prevalencia, desde 1911 hasta el año 1997, según revisión realizada por Ishida Yamamoto y colaboradores la literatura reporta solo treinta casos a escala mundial.
Respecto al caso de eritrokeratodermia.Una de mis primeras publicacione en el Bol.Hospital Carlos J Bello(ahora extinto)fué con una familia con este tipo de casos.Nosotros la llamamos erithroqueratodermia congenita simétrica.La publicacón fué en 1960 ó 61
Fugit irreparibile tempus
Mauricio Goihman