
Autores:
* Dra. Laura Mola Reyes, ** Dr. Ernesto Arteaga Hernández.
* Especialista de Primer Grado en Dermatología. Especialista de Primer Grado en Medicina General Integral. Máster en Urgencias Médicas en Atención Primaria de Salud. Hospital Hermanos Ameijeiras, La Habana, Cuba.
** Especialista de Segundo grado en Anatomía Patológica. Profesor auxiliar. Hospital Hermanos Ameijeiras, La Habana, Cuba.
Resumen
La Neurofibromatosis o enfermedad de Von Recklinghausen, es una genodermatosis autosómico dominante que se caracteriza clínicamente
por: manchas café con leche, tumores neurofibromatosos, efélides en axilas e ingles, nódulos de Lish y defectos óseos, neurológicos, viscerales, endocrinos y psiquiátricos. Presenta varios subtipos, entre ellos la neurofibromatosis segmentaria, que es un raro trastorno de clínica variable y de buen pronóstico, caracterizada por neurofibromas con o sin cambios pigmentarios, localizados con mayor frecuencia en una sola región del cuerpo
sin afectación visceral. Corresponde al tipo V de las neurofibromatosis según la clasificación de Ricardi y Eichner, es una afectación que predomina en mujeres en una relación de 2:1. Se reporta el caso de una paciente de 51 años de edad que acude a consulta porque presenta neurofibromas localizados en miembro superior derecho.
Palabras claves: Neurofibromatosis segmentaria verdadera.
Abstract
The Neurofibromatosis or Von Recklinghausen disease, is an autosomal dominant disorder that is characterized clinically by: coffee with milk stains, neurofibromas, ephelides in armpits and groins, Lish nodules and bone, neurological, visceral, endocrine and psychiatric defects, which has several subtypes, including that nerurofibromatosis segmental, a rare disorder clinic and variable good prognosis, characterized by neurofibromas with or without pigmentary changes, which are located more frequently in one region of the body without visceral involvement. It is for the V type of neurofibromatosis according to the classification of Ricardi and Eichner, it’s an affectation prevalent in women in a 2:1 relationship.
We report the case of a 51-year-old woman who consulted for neurofibromas in the right arm.
Key words: True Segmental Neurofibromatosis.
Introducción:
Las neurofibromatosis constituyen un grupo de enfermedades neurocutáneas, de herencia autosómica dominante que muestran extrema heterogeneidad clínica. Se caracterizan por crecimientos anormales en tejidos derivados de la cresta neural embriogénica que afectan la piel, los tejidos blandos, el sistema nervioso y los huesos. (1,2)
La neurofibromatosis clásica, que nace con el nombre de von Recklinghausen, comprende 85 a 90% de todos los casos de neurofibromatosis, ella es una enfermedad muy heterogénea donde un 10 a 15% corresponde a las variedades no clásicas. Dentro de la variedad no clásica encontramos la neurofibromatosis segmentaria (NFS). Schwartz y col. refieren que han sido reportados en la literatura aproximadamente 100 casos a nivel mundial, desde que fue descrita inicialmente por Crowe en 1956 como neurofibromatosis sectorial. (2,3)
Caso clínico
Paciente del sexo femenino, blanca, de 51 años de edad, que refiere que hace más de 15 años comenzaron a aparecer lesiones cutáneas nodulares del color de la piel, asintomáticas y de evolución progresiva en el miembro superior derecho.
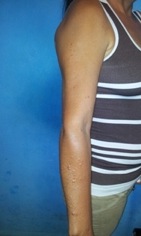
Al examen dermatológico, se observan, en la región externa del brazo y antebrazo derecho, nódulos sésiles de 0,5 mm a 10 mm de diámetro, de consistencia blanda, móviles no adheridos a planos profundos, de coloración semejante a la piel. Tienen una consistencia blanda y a la compresión tienden a invaginarse a través de un pequeño orificio en la piel (signo del ojal). Ausencia de efélides intertriginosas o manchas cafe-au-lait en el cuerpo (Ver Figura 1), ausencia de manifestaciones sistémicas, historia familiar negativa.
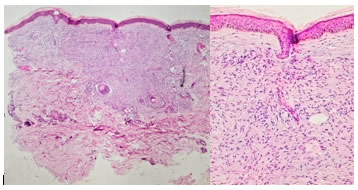
El estudio histopatológico (Figura 2) con hematoxilina y eosina, informó: Proceso proliferativo dérmico mesenquimatoso que no afecta la epidermis ni estructuras anexiales, con patrón fusocelular y fondo fibrilar sin atipia, compatible con Neurofibroma.
El diagnóstico definitivo fue de Neurofibromatosis segmentaria verdadera.
Discusión
En el año 1977 Miller y Sparkes introducen el término de segmentaria. En reconocimiento a la heterogeneidad de esta enfermedad, Riccardi en 1982 sugirió la división de la neurofibromatosis en 8 tipos. En esta clasificación, se designó la NF-V a la que presentaba neurofibromas limitados a un segmento del cuerpo y/o manchas café con leche, sin antecedentes familiar. (3)
La forma segmentaria fue revisada por Roth en 1987, y la dividió en cuatro subtipos, a la forma segmentaria de Riccardi la denominó de verdadera o subtipo 1, caracterizada como: unilateral, cutánea, y no hereditaria. (3)

Crowe estableció tres criterios para el diagnóstico de esta variedad:
1. Neurofibromas unilaterales limitados a uno o más nervios adyacentes.
2. Inexistencia de manchas café con leche limitadas a la región afectada.
3. Y la ocurrencia presumida de una mutación somática, con distribución esporádica y acentuada reducción o ausencia en la frecuencia de neurofibromatosis en las generaciones de los pacientes comprometidos. (3-6)
Se ha descrito que la forma segmentaria de la neurofibromatosis afecta predominantemente a las mujeres en una relación de 2:1 con los hombres. Edad de inicio es típicamente bimodal con picos entre los 10 a 30 años y 50 años. No se han reportado casos en el nacimiento. Los neurofibromas ocupan más comúnmente los dermatomas a nivel cervical o torácico y son unilaterales, ocurriendo más frecuentemente en el lado derecho que en el izquierdo. (3)
En un 6% de los casos se observa compromiso bilateral. Los sitios más afectados son: tórax y abdomen (55%), brazos (20%), miembros inferiores y cara (10%). Contrariamente a la NF tipo 1, el compromiso sistémico es infrecuente aunque según Hager et al podría llegar hasta el 21% de los casos.
Las manchas café con leche se observaron en aproximadamente un 26% de los casos con NFS, pudiendo presentarse también pecas, neurofibromas plexiformes y menos comúnmente nódulos de Lisch.(5)
Los neurofibromas en NFS son generalmente estructuras blandas, no dolorosas y raramente encapsuladas de difícil control postquirúrgico debido a la alta tasa de recurrencia a largo plazo y aunque son considerados uniformemente benignos se encuentran descriptos en la literatura casos que desarrollaron tumores malignos de nervios periféricos. (5,6)
La transmisión genética de la neurofibromatosis segmentaria es discutible. Crowe y Riccardi, admiten la hipótesis de mutación genética poscigótica en el inicio del desarrollo embrionario, en una célula de la cresta neural primitiva. Por las mitosis subsecuentes, un cierto número de células sería comprometido, motivando la distribución limitada de la enfermedad. La ocurrencia de lesiones bilaterales correspondería a una doble mutación somática. Hay casos descritos en los cuales la transmisión genética de NF-V, se da de un padre a su hijo y otro en que un paciente con NF-V tiene descendencia con neurofibromatosis clásica, se postula que el mecanismo genético se debería a que el mosaicismo somático acompañaría a un mosaicismo gonadal. Tinschert y col, mediante la técnica de hibridación con fluorescencia «in situ» demuestran que en la NF segmentaria ocurre una mutación del gen NF1 y que la distribución regional de las manifestaciones reflejaría clones celulares diferentes, correspondientes con el concepto de mosaicismo somático. Independientemente de la localización y del tamaño del área afectada del cuerpo los pacientes con NF-V estarán siempre en riesgo de mosaicismo y pueden por lo tanto transmitir el fenotipo, pero no el mosaico a la próxima generación. (3,7)
En la NFS con neurofibromas como manifestación única de la enfermedad, la aparición de los mismos sigue una distribución neural en dermatomas porque la mutación genética estaría limitada a las células de Schwann, mientras que aquellos con cambios pigmentarios solamente, la mutación ocurriría en los fibroblastos y la distribución de las lesiones seguiría las líneas cutáneas de Blaschko (no asociadas a estructuras nerviosas) (5)
Los neurofibromas son confirmados con el examen histopatológico respectivo. Al examen histopatológico de las lesiones, con hematoxilina-eosina, se encuentra habitualmente una lesión bien delimitada aunque no encapsulada; ocasionalmente los límites entre la lesión y el tejido circundante son imprecisos e infiltra el tejido conectivo dérmico. Los tumores están compuestos de fibras onduladas, delgadas, ligeramente eosinofílicas, formando bandas que se extienden en diversas direcciones y entre ellas se reconocen células con núcleos ovales o fusiformes, uniformes en tamaño. A veces se observa degeneración mucoide del estroma y en esas áreas los núcleos celulares están presentes en una sustancia homogénea, azul pálida consistente en mucopolisacáridos digeridos. (8)
Se ha considerado que los neurofibromas tienen buen pronóstico, sin riesgo de desarrollar una tumoración maligna en vaina nerviosa periférica, que es característico en los pacientes que tienen la forma generalizada de la enfermedad clásica. Las indicaciones para remoción quirúrgica de un neurofibroma en un paciente con neurofibromatosis incluyen: dolor, deterioro por compresión neurológica, compresión de estructuras adyacentes, desfiguración cosmética y crecimiento rápido sugestivo de degeneración maligna. Las indicaciones quirúrgicas son similares para pacientes con NF-V. Son referidos dos casos de malignidad en NF-V por Schwarz y col, los que concluyen que todos los pacientes con neurofibromas deberían ser considerados con un alto riesgo de degeneración maligna. (2,3,9)
El caso presentado cumple con los criterios exigidos para el encuadramiento en la forma segmentaria de la neurofibromatosis, por presentar exclusivamente neurofibromas unilaterales, disposición zosteriforme, sin antecedentes familiares de la enfermedad. En conclusión, el caso presentado cumple todos los criterios de una neurofibromatosis segmentaria, NF 5 de la clasificación de Riccardi, del subtipo 1, segmentaria verdadera, de acuerdo a la clasificación de Roth de la NF 5.
El interés de publicar el caso radica en que se trata de una entidad infrecuente y de difícil manejo debido a la alta tasa de recidiva de los neurofibromas, cuyo diagnóstico es importante para enfocar el tratamiento correcto y posterior seguimiento del caso, dado la transformación maligna que se ha descrito que pudiera ocurrir en estos casos.
Referencias bibliográficas:
1. Listernick R, Charrow J. The neurofibromatoses. In: Wolff K, Goldsmith L, Katz SI. Fitzpatrick’sdermatology in general medicine. 7ed. Nueva York: McGraw-Hill; 2008. p. 1331-39.
2. Fuente Rodríguez N, Tápanes Domínguez A, Pérez La O P. Neurofibromatosis tipo 1, enfermedad de Von Recklinhausen. Rev Cub Med Mil (serie en internet). 2007 Dic [citado 2010 Abr 7]36(4):(aprox. 5 p). Disponible en: http://scielo.sld.cu/scielo.php?script=sci_arttext pid=S0138-65572007000400009 & lng=es.
3. Vidarte G, ANADELA L, Ruiz E. Neurofibromatosis segmentaria verdadera. Dermatología peruana 2000; 10 (2):124-126.
4. Sezer E, Senayli A, Seazer T, Bicakci U. Segmental neurofibromatosis: Report of two cases. Journal of Dermatology. 2006; 33(9): 635-8.
5. Landriel F, Ferrara P, Hem S, Carrizo A.Neurofibromatosis segmentaria fronto témporo orbitaria: Reporte de un caso. Revisión de la literatura.Rev. argent. neurocir. v.21 n.3 Ciudad Autónoma de Buenos Aires jul./sep. 2007
6. Campollo Rodríguez I; Rodríguez Rojas JL; Limache Yaringaño LM. Neurofibromatosis segmentaria: presentación de un caso. AMC vol.15 no.6 Camagüey nov.-dic. 2011
7. Tinschert S, et al. Segmental neurofibromatosis is caused by somatic mutation the neurofibromatosis type 1 (NF1) gene. Eur J Hum Genet 2000; 8(6): 455-9.
8. Pascual Castroviejo I, Pascual Pascual S, Velázquez Fragua R, Viaño J, López Gutiérrez J. Neurofibromatosis segmentaria en niños: presentación de 43 pacientes. Rev Neurol. 2008;47(8):399-403.
9. Saettone-León A.Neurofibromatosis segmentaria. Reporte de un caso. Dermatol. Peru. v.16 n.1 Lima ene./abr. 2006
Hermoso Caso. Mil gracias por compartirlo. Volví a recordar esta patología.
Éxitos
Dra. Karina Alvarenga
Dermatóloga
Honduras