
SINDROME DE GARDNER Y ENFERMEDADES POLIPOSAS INTESTINALES ASOCIADAS A MUTACION GEN APC
GARDNER’S SYNDROME AND POLIPOID CONDITIONS ASOCIATED TO MUTATIONS IN THE APC GENE
Palabras clave: GARDNER- APC –SINDROME
Key words: GARDNER’S-APC-SYNDROME
Stefania Toso D.(1), Gabriela Morán C.(1), Isabel Jimeno O.(2), Rodrigo Loubies M.(3), Héctor Fuenzalida C.(3), Laura Segovia G.(4) Jaqueline Ampuero (5)
1-Residente Dermatología Universidad de Santiago de Chile, Servicio Dermatología Hospital El Pino
2- Médico Cirujano General Universidad de Santiago de Chile, Centro de Salud Familiar Salvador Allende Gossens, Santiago, Chile.
3- Dermatólogo Servicio de Dermatología Hospital El Pino, Santiago, Chile
4- Anatomo-Patológa, Hospital Barros Luco Trudeau, Santiago, Chile
5- Médico, Servicio de Endoscopía Hospital El Pino, Santiago, Chile
Resumen:
El Síndrome de Gardner corresponde a la asociación de pólipos colónicos adenomatosos, osteomas y tumores de tejido blando (quistes epidérmicos, fibromas y tumores desmoides). Fue descrita por Gardner en 1951 y se relacionaría mutaciones del gen APC.
La importancia de sospechar y diagnosticar un síndrome de Gardner de forma precoz sería evitar las complicaciones secundarias a las patologías asociadas tales como cáncer de colon, cáncer de tiroides, hepatoblastoma entre otros. Por lo que los quistes epidérmicos múltiples serían un marcador cutáneo de patología a tener en consideración.
Se describe un caso de un paciente de sexo masculino de 31 años con múltiples quistes en la piel y sin estudio previos.
Summary:
Gardner’s syndrome corresponds to the association of adenomatous colonic polyps, osteomas and soft tissue tumors (epidermal cysts, fibromas and desmoids tumors). It was first described by Gardner in 1951 and it is related to mutations in the APC gene.
The importance of suspecting and diagnosing promptly a Gardner’s Syndrome relies in treating and preventing the secondary complications such as colon cancer, thyroid cancer, hepatoblastoma and others. Thus multiples epidermal cysts would represent a cutaneous marker of disease that has to be considered by medical workers.
We describe a case of a male patient of 31 years old with multiple cysts in the skin and no previous studies.
Caso clínico:
Hombre de 31 años, consulta por múltiples quistes en piel. Sin estudios previos y con extirpación de dos quistes hace 1 año.
Refiere que dos hermanos tienen lesiones similares y madre fallecida por hemorragia gastrointestinal de etiología desconocida.
Al examen físico destacan múltiples lesiones quísticas de consistencia blanda con opérculo central, bien limitados, en dorso, abdomen, muslos, región cervical. Además presentaba algunas placas eritemato-escamosas en la zona de implantación del pelo y región abdominal y sacra.
Hematología general normal, colonoscopía con cientos a miles de pólipos, endocoscopía alta con pólipos aislados, ecotomografía tiroidea sin hallazgos y TAC de abdomen-pelvis con adenomas suprarrenales bilaterales. Se realiza biopsia excisional de tumores cutáneos con diagnostico histológico de quiste epidérmico. Con estos hallazgos se diagnostica Síndrome de Gardner.
Como conclusión, siempre sospechar genodermatosis o síndromes cuando se evidencian tumores cutáneos múltiples. Si son quísticos se debe realizar estudio gastrointestinal y manejo multidiciplinario de estos pacientes por el alto riesgo de cáncer colónico y patologías asociadas.

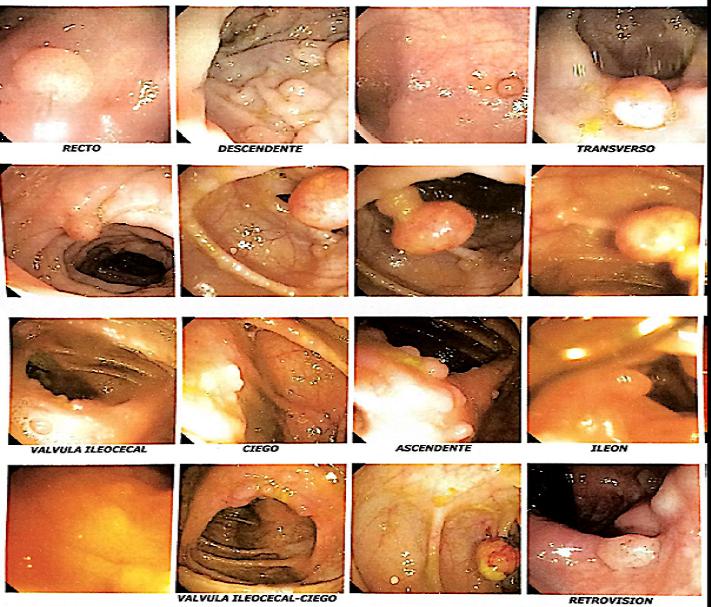
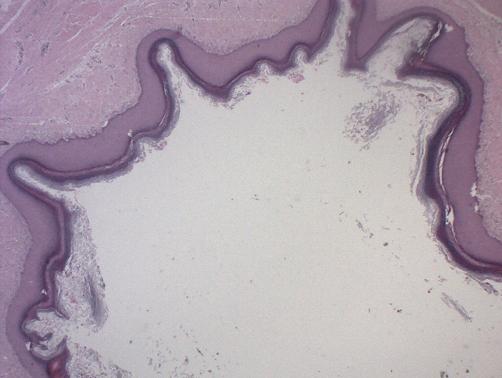
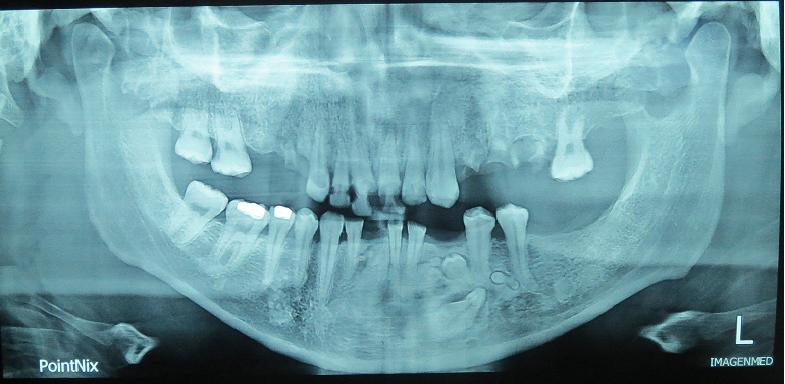
Discusión:
El gen APC es el único gen en el cual las variantes patológicas causan enfermedades poliposas asociadas a APC, siendo el diagnostico principalmente clínico (1). Los test de detección genética se realizan principalmente en el diagnostico temprano de los familiares en riesgo de tener estas enfermedades pero también para confirmar el diagnostico de PAF o poliposis adenomatosa familiar atenuada cuando los hallazgos de pólipos no cumplen con los criterios establecidos (menos de 100 pólipos adenomatosos).(2)
Los síndromes con mutación en la línea germinal en el gen APC (gen supresor tumoral), corresponden al Síndrome de Gardner, Síndrome de Turcot, Poliposis Adenomatosa Colónica Atenuada y Poliposis Adenomatosa Familiar. Muchas veces son indistinguibles y se sobreponen los fenotipos de estas entidades.(3,4)
La Poliposis Adenomatosa Familiar (PAF) Tiene una incidencia aproximada de 1 caso por cada 7500 recién nacidos vivos. Se caracteriza por la presencia de múltiples pólipos adenomatosos de gran tamaño en el intestino durante la infancia y adolescencia y el desarrollo inevitable de cáncer colorrectal. Se estima que para la edad de 35 años 95% de los individuos tiene pólipos. La edad promedio de detección de cáncer colónico en pacientes sin tratamiento es de 39 años.(5,6) Se describe que los pacientes con PAF tienen un riesgo entre 750 a 7500 veces mayor de desarrollar un hepatoblastoma que la población general, siendo la mayoría de estos previos a la edad de 3 años.
En cambio la Poliposis Adenomatosa Colónica Atenuada correspondería a una forma atenuada de PAF en la cual los pacientes desarrollarían menor cantidad de adenomas a una edad más tardía y tendrían un menor riesgo de cáncer. (7)
El Síndrome de Turcot es la asociación de poliposis colónica y tumores del sistema nervioso central, usualmente meduloblastoma.
El Síndrome de Gardner, tiene una incidencia aproximada de 1 en 4000 o 1 en 12000, se caracteriza por pólipos colónicos típicos de la PAF junto con osteomas y tumores de tejido blando. Es un síndrome raro, autosómico dominante el cual tiene un buen pronostico si se logra detectar a tiempo y se maneja de manera multidisciplinaría.(8)
Dentro de las anormalidades esqueléticas se describen los osteomas, que son tumores benignos de crecimiento lento y continuo. Ocurriendo de manera más frecuente en la mandíbula, la corteza externa de la calota y en los senos paranasales. Siendo el ángulo de la mandíbula un sitio típico. Por lo que encontrar en una radiografía panorámica más de tres de estas lesiones sería altamente sugerente de Síndrome de Gardner. Se describe que entre un 10-30% de los pacientes con PAF desarrollan estas lesiones y que el riesgo es 800 veces mas que en la población general.(9)
Cuando se analizan las anormalidades dentales se puede apreciar que existen hasta en un 30% de los pacientes, describiéndose dientes supernumerarios, odontomas compuestos, hipodontia, morfología dentaria anormal y dientes que aun no erupcionan.
Al analizar las lesiones cutáneas se describe que hasta un 65% de los pacientes tiene múltiples quistes epidérmicos, que estos aparecerían antes de la pubertad y que ocurren primariamente en la cara, cuero cabelludo y extremidades. Al contario de los tumores desmoides que pueden aparecer en la piel de la pared abdominal anterior o de manera intra-abdominal, Ocurriendo en un 80% de los pacientes con PAF para la edad de 40 años (10). Estos tumores pueden comprimir órganos abdominales, siendo descrito que hasta un 5% de los pacientes con PAF experimentan morbilidad y o mortalidad por estos tumores.
En relación a las masas adrenales se describe en los pacientes con PAF son 2 a 4 veces mas prevalentes que en la población general. Siendo la mayoría adenomas asintomáticos (11)
Conclusión:
Al ser las enfermedades generadas por mutaciones del gen APC, muchas veces indistinguibles y con entidades que se sobreponen se debe tener en consideración que las comorbilidades y complicaciones pueden ser experimentadas por los diferentes síndromes, no siendo exclusivas de cada uno. Es decir que al enfrentar un paciente con múltiples quistes epidérmicos debemos sospechar que puede tener comorbilidades asociadas como pólipos y por lo tanto riesgo de cáncer de colon, osteomas y anormalidades dentales, tumores de tejido blando entro otros. Por lo que la sospecha será de vital importancia y el poder evaluarlo de manera multidisciplinaría para evitar las complicaciones y la detección tardía de estas es lo principal.
Más aún al conocer que los pacientes con PAF tienen superposición de patologías con las enfermedades asociadas a la mutación de APC, se podrán evaluar de manera correcta por el dermatólogo, al tener en consideración las lesiones coexistentes que pueden presentar.
Nuestro paciente esta siendo evaluado de manera multidisciplinaría por los cirujanos maxilo-faciales, dentistas, cirujano digestivo bajo, gastroenterólogo, dermatólogo, sicólogo y medicina interna. Se ha diferido la colectomía total ya que el paciente rechazó la cirugía por motivos personales.
Referencias
- Gardner RJ, Kool D, Edkins E, et al. The clinical correlates of a 3′ truncating mutation (codons 1982-1983) in the adenomatous polyposis coli gene. Gastroenterology. Jul 1997;113(1):326-31.
- Debinski HS, Love S, Spigelman AD, et al. Colorectal polyp counts and cancer risk in familial adenomatous polyposis. Gastroenterology. Apr 1996;110(4):1028-30
- Burt RW, Ward K, Spirio L. et al. Accurate identification of familial adenomatous polyposis coli using newly developed genetic markers. Gastroenterology. 1992;102:A347.
- Juhn E, Khachemoune A. Gardner syndrome: skin manifestations, differential diagnosis and management. Am J Clin Dermatol. 2010;11(2):117-22.
- EJ, Richard RC. Multiple cutaneous and subcutaneous lesions occurring simultaneously with hereditary polyposis and osteomatosis. Am J Hum Genet. 1953;5:139–147.
- Tulchinsky H, Keidar A, Strul H, et al. Extracolonic manifestations of familial adenomatous polyposis after proctocolectomy. Arch Surg. Feb 2005;140(2):159-63; discussion 164.Burt RW, Leppert MF, Slattery ML, Samowitz WS, Spirio LN, Kerber RA, Kuwada SK, Neklason DW, Disario JA, Lyon E, Hughes JP, Chey WY, White RL. Genetic testing and phenotype in a large kindred with attenuated familial adenomatous polyposis. Gastroenterology. 2004;127:444–51.
- Karazivan, M., Manoukian, K., and Lalonde, B.:”Familial adenomatous polyposis or Gardner syndrome review of the literature and presentation of 2 clinical cases”, J Can Dent Assoc, 2000 Jan; 66 (1): 26-30.
- Takeuchi T, Takenoshita Y, Kubo K, Iida M. Natural course of jaw lesions in patients with familiar adenomatosis coli (Gardner’s syndrome) Int J Oral Maxillofac Surg. 1993;22:226–230.
- Bhama PK, Chugh R, Baker LH, Doherty GM. Gardner’s syndrome in a 40-year-old women: successful treatment of locally aggressive desmoid tumor with cytotoxic chemotheraphy. World J Surg Oncology. 2006;4:90–100.
- Smith TG, Clark SK, Katz DE, Reznek RH, Phillips RK. Adrenal masses are associated with familial adenomatous polyposis. Dis Colon Rectum. 2000b;43:1739–42.
Muy bonito caso y excelente discusión. Gracias por compartir.
Excelente presentación, me gustaría saber si se evaluó el fondo de ojo de este paciente por si existiera concordancia con hipertrofia congénita del epitelio pigmentario de la retina. Gracias.
Buen caso, bien estudiado y presentado. Gracias por compartir